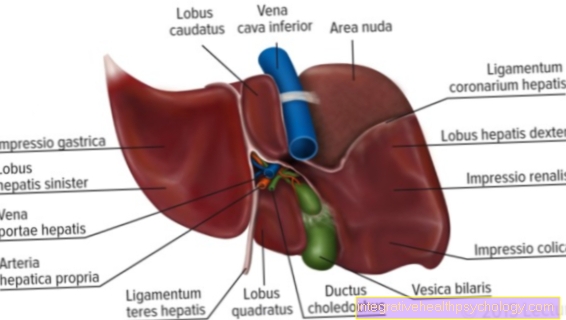
гемофилия
Синонимы
Гемофилия, наследственное нарушение свертываемости крови, дефицит фактора свертывания крови,
Дефицит фактора VIII, дефицит фактора IX, гемофилия
определение
Гемофилия - наследственное заболевание системы свертывания крови:
У пораженных пациентов наблюдается нарушение свертываемости крови, что проявляется в том, что кровотечение происходит при малейшей травме и кровотечение трудно остановить.
Факторы свертывания не могут быть активированы, поэтому регулярное протекание каскада коагуляции невозможно.
Гемофилия - это врожденное заболевание, которое бывает двух форм:
Может присутствовать гемофилия A и B.
Гемофилия A встречается чаще (в 85% случаев), чем форма B (примерно 15%), и является более серьезной из двух форм.
Активность комплекса фактора VIII ограничена при гемофилии А, потому что образование фактора VIII-C в клетках печени снижено.
Фактор VIII-C является одной из двух субъединиц комплекса фактора VIII: фон-Виллебранда-Фактор и фактор VIII-C образуют этот комплекс, способствующий свертыванию, в каскаде коагуляции. Фактор фон Виллебранда защищает фактор VIII-C от слишком быстрого расщепления.
Следовательно, количество функционального комплекса фактора VIII снижается в форме А.
У которой гемофилия B. синтез фактора свертывания IX происходит в меньшей степени, поэтому его влияние на процесс свертывания отсутствует и гемостаз нарушается.
Возникновение / эпидемиология
Встречаемость гемофилии среди населения относится к гемофилии А. 1 из 5000 а для формы Б. 1:15.000.
Тот факт, что мужчины особенно болеют, связан с гендерными проблемами.
Х-сцепленная форма наследования дефекта коагуляции (см. Причина / развитие).
основы
Свертывание крови - это точно настроенная система: различные компоненты крови, так называемые факторы свертывания, активируются один за другим каскадным образом, чтобы обеспечить гемостаз в случае травмы.
Система коагуляции состоит из первичного и вторичного гемостаза, который также называется плазматической коагуляцией.
Если стенка сосуда повреждена, пораженный сосуд на первом этапе сужается (= первичная коагуляция). Тогда опора (=Тромб тромбоцитов) на травмированном участке, покрытом тромбоцитами (= Тромбоциты) формируется для достижения начальной герметичности сосуда.
Образование тромба активирует плазматическую коагуляцию:
Начинается каскад коагуляции, и взаимодействие различных факторов свертывания создает стабильное закрытие раны или сосудов.
Причина и происхождение
гемофилия Обе формы гемофилии наследуются как Х-сцепленный рецессивный признак.
При так называемом X-сцепленном способе наследования гемофилии генетическая информация для продукта гена находится на половой хромосоме X, которая содержится в двойных числах у женщин и в единичных числах у мужчин.
Для каждой генетической информации существует два гена, так называемые аллели. Если имеется рецессивное наследование, оба аллеля должны быть затронуты мутацией, чтобы вызвать заболевание: рецессивный означает, что патологически измененная генетическая информация получена от здорового, т.е. не меняется, информация подавляется и не приводит к болезни. Рецессивное наследственное заболевание развивается только в том случае, если оба аллеля несут неверную генетическую информацию.
Только Выражение гена изменилось, да ладно Не в гемофилия.
Информация: наследование
Если изменить экспрессию только одного гена, болезни не будет.
Поскольку мужчины имеют только одну Х-хромосому в паре половых хромосом и, следовательно, информация о генных продуктах на их Х-хромосоме доступна только в простой форме, они заболеют, если будет дефектен только один аллель.
С другой стороны, женщины, имеющие две Х-хромосомы в качестве половых хромосом, заболевают только тогда, когда обе хромосомы имеют дефектные гены для генного продукта.
У мужчин, страдающих гемофилией, аллель, несущий наследственную информацию для фактора свертывания крови VIII-C (гемофилия A) или фактора свертывания крови IX (гемофилия B), доступен только в слегка измененной форме;
Чтобы заболеть, женщины должны нести это генетическое изменение на обоих аллелях.
Если у женщины есть дефектный и неизмененный аллель, их называют носителями Х-сцепленного наследственного заболевания, такого как гемофилия.
При гемофилии ложная генетическая информация снижает активность пораженного фактора свертывания или снижает выработку фактора свертывания крови.
Х-хромосома является носителем генетического материала факторов свертывания крови для вторичного гемостаза.
Прибл. 70% мутаций в генетическом материале, вызывающих гемофилию, передаются в семьях, 30% заболеваний возникают в результате спонтанных изменений (= спонтанных мутаций) в генетической информации.
диагностика
После того, как вы спросите об истории болезни пациента и проведете тщательное физическое обследование, есть дополнительные шаги в диагностике гемофилии:
В 2/3 случаев есть случаи гемофилии в семье, поэтому следует спросить о семейной кластеризации болезни, если пациент обращается к врачу с симптомами, подозрительными на гемофилию.
Пациенты сообщают о синяках в результате самых мелких травм.
Различают те Степень тяжести гемофилии в легкую, среднетяжелую или тяжелую форму заболевания.
Исследование образца крови дает следующие результаты, типичные для гемофилии:
Время кровотечения нормальное (= первичная коагуляция не нарушена), но функция плазматической коагуляции снижена, поэтому так называемое время PTT больше.
ЧТТ обозначает частичное тромбопластиновое время. Он измеряется в лабораторных тестах для проверки функции факторов I, II, V, VIII-XII, а также XIV и XV.
Поскольку при гемофилии отсутствует один фактор в каскаде коагуляции, он не может работать оптимальным образом, что приводит к увеличению времени коагуляции.
Для того чтобы различать гемофилию А и В, кровь пациента необходимо исследовать на факторы VIII и IX; отсутствие этих двух факторов определяет тип гемофилии.
Дифференциальная диагностика
Гемофилию следует дифференцировать от других нарушений свертывающей системы, в частности, упоминая синдром фон Виллебранда-Юргенса.
Фактор фон Виллебранда (vWF) работает вместе с фактором VIII-C im.
Фактор VIII является комплексным и вызывает ингибирование преждевременной деградации фактора VIII, кроме того, фактор способствует коагуляции, опосредуя адгезию тромбоцитов крови к поврежденному участку сосуда. Таким образом, vWF является важной частью как первичной, так и вторичной коагуляции.
Если синдром фон Виллебранда-Юргенса присутствует, то формирование
vWFs в клетках стенки сосуда имеют место только в небольшой степени или неправильным образом. Сниженный уровень образования или неадекватная функция
Фактор фон Виллебранда вызывается мутациями в гене образования фактора. Наследование синдрома или мутации причинного гена наследуется по аутосомно-доминантному типу: генетическая информация для vWF находится на хромосоме номер 12, которая не является половой хромосомой. При доминантном способе наследования больной аллель подавляет действие второго, здорового аллеля, так что болезнь вызвана мутацией гена и вызывает клинические симптомы.
Пациенты обычно страдают от кровотечений на коже и слизистых оболочках, реже от спонтанных кровотечений, как у пациентов с гемофилией.
Заболевание часто замечают только при длительном кровотечении после операции.
У больного после травмы время кровотечения увеличивается, а значения плазматической коагуляции патологически изменяются (пролонгированная ЧТВ).
Синдром фон Виллебранда-Юргенса вызывается введением десмопрессина или заменой фактора VIII и vWF для стабилизации коагуляции (более подробное объяснение см. В разделе «Терапия гемофилии»).
Какие симптомы гемофилии?
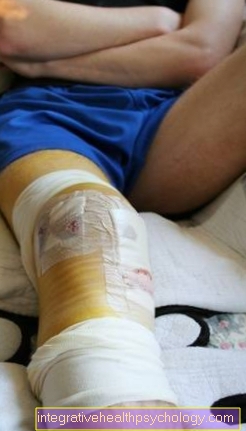
Симптомы при формах гемофилии не отличаются:
- Гемофилия обычно приводит только к клиническим симптомам у мужчин.
- Чрезмерное кровотечение возникает при гемофилии, что не имеет отношения к предыдущему, в основном банальному происшествию (травма), время кровотечения нормальное, но обычно возникает кровотечение, которого не бывает у здоровых людей.
- Различают три формы гемофилии, которые определяются остаточной активностью пораженных факторов свертывания крови:
1.тяжелая гемофилия (гемофилия) с остаточной активностью менее 1% или оставшейся долей функционального фактора, при котором происходит спонтанное кровотечение и кровотечение суставов
2. Умеренная гемофилия (заболевание крови) с активностью фактора в диапазоне от
1 и 5% нормальной активности фактора и появление синяков (= гематом) после незначительной травмы
3. легкая гемофилия (гемофилия), при котором остаточная активность все еще составляет 5–15%, и пациенты сообщают о таких симптомах, как гематомы (синяки) после значительной травмы и послеоперационного кровотечения.
Подробнее по теме: Каковы причины кровоизлияния в мозг
- Женщины, у которых обычно имеется только измененная генетическая информация о гемофилии, обычно не проявляют клинических симптомов с остаточной активностью более 50%.
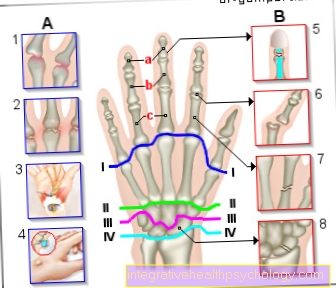
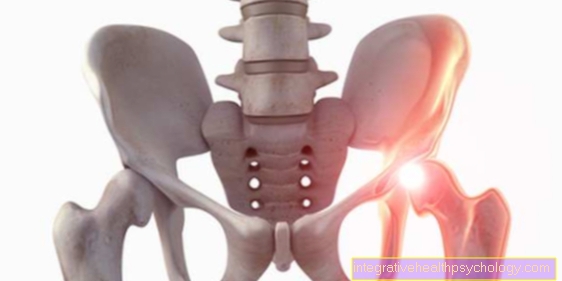
- Пациенты испытывают подобное кровотечение в крупных суставах колено-, тазобедренный- или Плечевой сустав, при этом говорят о гемартрозе. Кровотечение вызывает воспалительную реакцию с восстановительными процессами в суставе, что может привести к его жесткости.
- Он также может кровоточить в мускулатура и наступают мягкие ткани. Кровотечение приводит к увеличению давления в мышцах или тканях, поскольку теперь объем увеличивается. Это может особенно повлиять на ваши руки и ноги. Компартментный синдром вести:
Повышенное давление вызывает сжатие кровеносных сосудов и нервов, в результате чего конечности получают недостаточное питание, и большие участки тканей могут погибнуть. Синдром компартмента необходимо лечить как можно скорее, чтобы предотвратить потерю конечности. - Возникает кровотечение в брюшную полость, что представляет опасность для жизни пациента.
- После операции возможно длительное кровотечение.
Кроме того, могут быть длительные периоды с кровью в моче. Здесь один угрожает малокровиепотому что пациент постоянно теряет кровь, возможно, незаметно. - Особенно опасны Кровоизлияние в мозг (= внутричерепное кровотечение), которое приводит к смерти у 10% людей с гемофилией. Вы можете узнать больше по этой теме в нашей теме Кровоизлияние в мозг.
Как проводится терапия гемофилии?

У больного должно возникнуть кровотечение гемофилия следует избегать, поэтому пациент не Медикамент которые препятствуют свертыванию крови, такие как Ацетилсалициловая кислота (Аспирин®) и отсутствие внутримышечных инъекций (= шприцы в мускулатура) соответственно.
В случае травмы с кровотечением большое значение имеет тщательный местный гемостаз, чтобы предотвратить кровотечение в соседние ткани.
Медикаментозная терапия гемофилия заключается в замене факторов свертывания, которые организм не может вырабатывать в достаточном количестве.
Пациенты с легким гемофилия получать препараты фактора свертывания крови по мере необходимости, т.е. если была травма с кровотечением или если планируется серьезная операция, которая может привести к обильному кровотечению.
Профилактическую замену фактора следует использовать у пациентов с гемофилией средней и тяжелой степени, т.е. недостающий коэффициент следует указывать постоянно, перед происходит кровотечение.
Если остаточная активность фактора VIII более 15% или фактора IX более 20-25%, регулярная терапия не требуется; эти пациенты получают недостающий фактор свертывания в случае спонтанного кровотечения или перед плановой операцией.
Долгосрочная терапия, а также терапия перед операцией может принимать форму домашнего самолечения, при котором пациент сам применяет недостающий фактор свертывания крови.
Для пациентов с легкой формой гемофилии существует альтернатива терапии:
Действующее вещество десмопрессин (например, Минирин®) приводит к распределению
Фактор VIII, который накапливается в стенках сосудов.
Однако лекарство можно давать только в течение нескольких дней, так как после стимуляции высвобождения фактора память в стенках сосудов истощается и ее необходимо воспроизводить снова.
При острой терапии гемофилия есть три варианта вмешательства:
- Пациент получает активированный протромбин, вещество, которое способствует свертыванию крови, так что кровь останавливается как можно быстрее.
- Другой терапевтический вариант - введение препаратов фактора VII. Этот фактор находится в начале каскада коагуляции и инициирует его регулярное течение.
- Третий терапевтический вариант - это применение животного фактора VIII-C для остановки кровотечения у пациента.
осложнения
Замещающий подарок (=замена) факторов свертывания крови может приводить к образованию антител именно против этих факторов, так что замена в постоянной дозе больше не имеет терапевтических эффектов, поэтому говорят об образовании ингибиторов.
В зависимости от определения концентрации антител в крови пациента могут вводиться высокие дозы фактора VIII, цель которых - восстановить толерантность к этому фактору и остановить образование антител. Эта процедура должна выполняться только в специализированных центрах.
Низкий риск заражения гепатитом или вирусом ВИЧ (=ВИЧ), поскольку факторы получены из продуктов крови человека.
Еще один риск заместительной терапии гемофилии - возникновение аллергических реакций.